bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
164,496 sequences; 82,071,388 total letters
Query= Eten_3405_orf2
Length=132
Score E
Sequences producing significant alignments: (Bits) Value
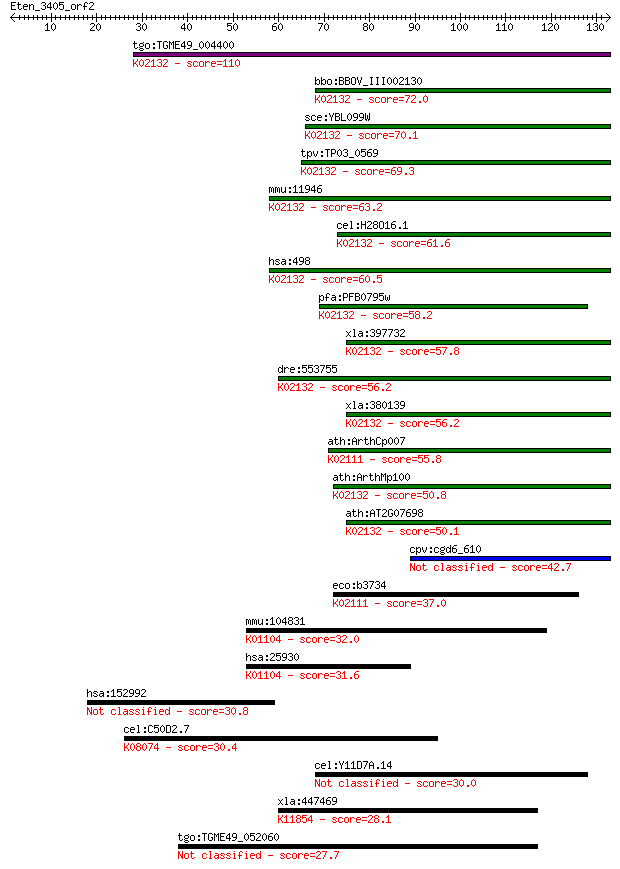
tgo:TGME49_004400 ATP synthase alpha chain, putative (EC:3.4.2... 110 1e-24
bbo:BBOV_III002130 17.m07204; ATP synthase F1, alpha subunit f... 72.0 5e-13
sce:YBL099W ATP1; Alpha subunit of the F1 sector of mitochondr... 70.1 2e-12
tpv:TP03_0569 ATP synthase F1 subunit alpha (EC:3.6.3.14); K02... 69.3 3e-12
mmu:11946 Atp5a1, AI035633, AL022851, AL023067, Atpm, D18Ertd2... 63.2 2e-10
cel:H28O16.1 hypothetical protein; K02132 F-type H+-transporti... 61.6 7e-10
hsa:498 ATP5A1, ATP5A, ATP5AL2, ATPM, MOM2, OMR, ORM, hATP1; A... 60.5 1e-09
pfa:PFB0795w ATP synthase F1, alpha subunit, putative; K02132 ... 58.2 7e-09
xla:397732 atp5a1, MGC84051, atp5a; ATP synthase, H+ transport... 57.8 9e-09
dre:553755 atp5a1, zgc:154103; ATP synthase, H+ transporting, ... 56.2 3e-08
xla:380139 atp5a1, MGC64338; ATP synthase, H+ transporting, mi... 56.2 3e-08
ath:ArthCp007 atpA; ATP synthase CF1 alpha chain; K02111 F-typ... 55.8 4e-08
ath:ArthMp100 atp1; ATPase subunit 1; K02132 F-type H+-transpo... 50.8 1e-06
ath:AT2G07698 ATP synthase alpha chain, mitochondrial, putativ... 50.1 2e-06
cpv:cgd6_610 ATP synthase alpha chain 42.7 3e-04
eco:b3734 atpA, ECK3727, JW3712, papA, uncA; F1 sector of memb... 37.0 0.017
mmu:104831 Ptpn23, AI462446, PTP-TD14; protein tyrosine phosph... 32.0 0.46
hsa:25930 PTPN23, DKFZp564F0923, HD-PTP, HDPTP, KIAA1471, PTP-... 31.6 0.70
hsa:152992 METTL19, C4orf23, FLJ12891, FLJ35725; methyltransfe... 30.8 1.1
cel:C50D2.7 hypothetical protein; K08074 ADP-dependent glucoki... 30.4 1.6
cel:Y11D7A.14 hypothetical protein 30.0 1.7
xla:447469 usp38, MGC81730; ubiquitin specific peptidase 38 (E... 28.1 7.7
tgo:TGME49_052060 hypothetical protein 27.7 9.0
> tgo:TGME49_004400 ATP synthase alpha chain, putative (EC:3.4.21.68);
K02132 F-type H+-transporting ATPase subunit alpha
[EC:3.6.3.14]
Length=565
Score = 110 bits (275), Expect = 1e-24, Method: Compositional matrix adjust.
Identities = 63/110 (57%), Positives = 74/110 (67%), Gaps = 6/110 (5%)
Query 28 ACAARLSARLPAAAAAAAGP-----AAAAGQQQQRRLLHTSDLRAAATTISPSEMAKILE 82
+C AR + + A+ A A A A G Q RLLHTS LRAA ISPSEM+++LE
Sbjct 5 SCLARRAVSVAASGARAFASGLGARAVAVGALQSARLLHTSSLRAAGAKISPSEMSRLLE 64
Query 83 QRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
+RI+GW TQ ST EVGRV SVGDGIARL GL V+AGELVE Q+G GM
Sbjct 65 ERIAGWKTQTSTE-EVGRVVSVGDGIARLFGLEGVQAGELVEFQNGMTGM 113
> bbo:BBOV_III002130 17.m07204; ATP synthase F1, alpha subunit
family protein; K02132 F-type H+-transporting ATPase subunit
alpha [EC:3.6.3.14]
Length=544
Score = 72.0 bits (175), Expect = 5e-13, Method: Compositional matrix adjust.
Identities = 33/65 (50%), Positives = 46/65 (70%), Gaps = 1/65 (1%)
Query 68 AATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQS 127
+ + ISP+EM+K+L+ RI+GW Q+ +VG V +VGDGIAR+ GL +V+ GELV S
Sbjct 30 STSKISPAEMSKLLQSRIAGWEKQKDVV-DVGHVINVGDGIARVYGLKDVKMGELVVFSS 88
Query 128 GAVGM 132
G GM
Sbjct 89 GVSGM 93
> sce:YBL099W ATP1; Alpha subunit of the F1 sector of mitochondrial
F1F0 ATP synthase, which is a large, evolutionarily conserved
enzyme complex required for ATP synthesis; phosphorylated
(EC:3.6.3.14); K02132 F-type H+-transporting ATPase subunit
alpha [EC:3.6.3.14]
Length=545
Score = 70.1 bits (170), Expect = 2e-12, Method: Compositional matrix adjust.
Identities = 34/67 (50%), Positives = 46/67 (68%), Gaps = 1/67 (1%)
Query 66 RAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVEL 125
R A+T P+E++ ILE+RI G + ++ E GRV +VGDGIAR+ GL N++A ELVE
Sbjct 34 RLASTKAQPTEVSSILEERIKG-VSDEANLNETGRVLAVGDGIARVFGLNNIQAEELVEF 92
Query 126 QSGAVGM 132
SG GM
Sbjct 93 SSGVKGM 99
> tpv:TP03_0569 ATP synthase F1 subunit alpha (EC:3.6.3.14); K02132
F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=545
Score = 69.3 bits (168), Expect = 3e-12, Method: Compositional matrix adjust.
Identities = 34/69 (49%), Positives = 48/69 (69%), Gaps = 2/69 (2%)
Query 65 LRAAATT-ISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELV 123
LR +T +SP+E++K+L+ +I GW +Q+ +VG V +VGDGIAR+ GL V+AGELV
Sbjct 29 LRPFSTNKVSPAEISKLLQTKIGGWDSQKDVR-DVGHVINVGDGIARIYGLKEVKAGELV 87
Query 124 ELQSGAVGM 132
SG GM
Sbjct 88 MFSSGVAGM 96
> mmu:11946 Atp5a1, AI035633, AL022851, AL023067, Atpm, D18Ertd206e,
Mom2; ATP synthase, H+ transporting, mitochondrial F1
complex, alpha subunit 1; K02132 F-type H+-transporting ATPase
subunit alpha [EC:3.6.3.14]
Length=553
Score = 63.2 bits (152), Expect = 2e-10, Method: Compositional matrix adjust.
Identities = 38/75 (50%), Positives = 46/75 (61%), Gaps = 3/75 (4%)
Query 58 RLLHTSDLRAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNV 117
R LH S+ R T +EM+ ILE+RI G T E GRV S+GDGIAR+ GL NV
Sbjct 34 RNLHASNTRLQKT--GTAEMSSILEERILGADTSVDLE-ETGRVLSIGDGIARVHGLRNV 90
Query 118 RAGELVELQSGAVGM 132
+A E+VE SG GM
Sbjct 91 QAEEMVEFSSGLKGM 105
> cel:H28O16.1 hypothetical protein; K02132 F-type H+-transporting
ATPase subunit alpha [EC:3.6.3.14]
Length=538
Score = 61.6 bits (148), Expect = 7e-10, Method: Compositional matrix adjust.
Identities = 32/60 (53%), Positives = 42/60 (70%), Gaps = 1/60 (1%)
Query 73 SPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
S SE++KILE+RI G T + E G+V S+GDGIAR+ GL N++A E+VE SG GM
Sbjct 32 SGSEVSKILEERILGTETGINLE-ETGKVLSIGDGIARVYGLKNIQAEEMVEFDSGIKGM 90
> hsa:498 ATP5A1, ATP5A, ATP5AL2, ATPM, MOM2, OMR, ORM, hATP1;
ATP synthase, H+ transporting, mitochondrial F1 complex, alpha
subunit 1, cardiac muscle; K02132 F-type H+-transporting
ATPase subunit alpha [EC:3.6.3.14]
Length=553
Score = 60.5 bits (145), Expect = 1e-09, Method: Compositional matrix adjust.
Identities = 36/75 (48%), Positives = 44/75 (58%), Gaps = 3/75 (4%)
Query 58 RLLHTSDLRAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNV 117
R H S+ T +EM+ ILE+RI G T E GRV S+GDGIAR+ GL NV
Sbjct 34 RNFHASNTHLQKT--GTAEMSSILEERILGADTSVDLE-ETGRVLSIGDGIARVHGLRNV 90
Query 118 RAGELVELQSGAVGM 132
+A E+VE SG GM
Sbjct 91 QAEEMVEFSSGLKGM 105
> pfa:PFB0795w ATP synthase F1, alpha subunit, putative; K02132
F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=551
Score = 58.2 bits (139), Expect = 7e-09, Method: Composition-based stats.
Identities = 29/59 (49%), Positives = 41/59 (69%), Gaps = 1/59 (1%)
Query 69 ATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQS 127
+T ISP E++KILE++ + + S+ EVG V SVGDGI R GL NV++ ELVE+ +
Sbjct 33 STKISPIEISKILEKKFESFNFKTSS-NEVGYVLSVGDGICRAYGLNNVKSSELVEIHN 90
> xla:397732 atp5a1, MGC84051, atp5a; ATP synthase, H+ transporting,
mitochondrial F1 complex, alpha subunit 1, cardiac muscle;
K02132 F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=553
Score = 57.8 bits (138), Expect = 9e-09, Method: Compositional matrix adjust.
Identities = 31/58 (53%), Positives = 39/58 (67%), Gaps = 1/58 (1%)
Query 75 SEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
+E++ ILE+RI G T E GRV S+GDGIAR+ GL NV+A E+VE SG GM
Sbjct 49 AEVSSILEERILGADTTADLE-ETGRVLSIGDGIARVYGLRNVQAEEMVEFSSGLKGM 105
> dre:553755 atp5a1, zgc:154103; ATP synthase, H+ transporting,
mitochondrial F1 complex, alpha subunit 1, cardiac muscle;
K02132 F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=551
Score = 56.2 bits (134), Expect = 3e-08, Method: Compositional matrix adjust.
Identities = 34/73 (46%), Positives = 44/73 (60%), Gaps = 3/73 (4%)
Query 60 LHTSDLRAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRA 119
LHT+ R +E++ ILE++I G T E GRV S+GDGIAR+ GL NV+A
Sbjct 35 LHTA--RPWLQKTGTAEVSSILEEKILGADTGAELE-ETGRVLSIGDGIARVYGLRNVQA 91
Query 120 GELVELQSGAVGM 132
E+VE SG GM
Sbjct 92 EEMVEFSSGLKGM 104
> xla:380139 atp5a1, MGC64338; ATP synthase, H+ transporting,
mitochondrial F1 complex, alpha subunit, isoform 1; K02132 F-type
H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=553
Score = 56.2 bits (134), Expect = 3e-08, Method: Compositional matrix adjust.
Identities = 30/58 (51%), Positives = 38/58 (65%), Gaps = 1/58 (1%)
Query 75 SEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
+E++ ILE+RI G E GRV S+GDGIAR+ GL NV+A E+VE SG GM
Sbjct 49 AEVSSILEERILGADISTDLE-ETGRVLSIGDGIARVYGLRNVQAEEMVEFSSGLKGM 105
> ath:ArthCp007 atpA; ATP synthase CF1 alpha chain; K02111 F-type
H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=507
Score = 55.8 bits (133), Expect = 4e-08, Method: Compositional matrix adjust.
Identities = 27/62 (43%), Positives = 38/62 (61%), Gaps = 1/62 (1%)
Query 71 TISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAV 130
TI E++ I+ +RI + ++ T G V VGDGIAR+ GL V AGELVE + G +
Sbjct 3 TIRADEISNIIRERIEQYN-REVTIVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGTI 61
Query 131 GM 132
G+
Sbjct 62 GI 63
> ath:ArthMp100 atp1; ATPase subunit 1; K02132 F-type H+-transporting
ATPase subunit alpha [EC:3.6.3.14]
Length=507
Score = 50.8 bits (120), Expect = 1e-06, Method: Compositional matrix adjust.
Identities = 26/63 (41%), Positives = 38/63 (60%), Gaps = 3/63 (4%)
Query 72 ISP--SEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGA 129
+SP +E+ + E RI + E+GRV SVGDGIA++ GL ++AGE+V +G
Sbjct 3 LSPRAAELTNLFESRIRNFYAN-FQVDEIGRVVSVGDGIAQVYGLNEIQAGEMVLFANGV 61
Query 130 VGM 132
GM
Sbjct 62 KGM 64
> ath:AT2G07698 ATP synthase alpha chain, mitochondrial, putative;
K02132 F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=777
Score = 50.1 bits (118), Expect = 2e-06, Method: Compositional matrix adjust.
Identities = 24/58 (41%), Positives = 35/58 (60%), Gaps = 1/58 (1%)
Query 75 SEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
+E+ + E RI + E+GRV SVGDGIA++ GL ++AGE+V +G GM
Sbjct 278 AELTNLFESRIRNFYAN-FQVDEIGRVVSVGDGIAQVYGLNEIQAGEMVLFANGVKGM 334
> cpv:cgd6_610 ATP synthase alpha chain
Length=639
Score = 42.7 bits (99), Expect = 3e-04, Method: Composition-based stats.
Identities = 20/44 (45%), Positives = 28/44 (63%), Gaps = 0/44 (0%)
Query 89 TTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVELQSGAVGM 132
T + +G+V SV DGIA++ G+ +V+ GELVE SG GM
Sbjct 141 TIHEDNNKRIGQVISVADGIAQVDGIRSVKYGELVEFSSGEKGM 184
> eco:b3734 atpA, ECK3727, JW3712, papA, uncA; F1 sector of membrane-bound
ATP synthase, alpha subunit (EC:3.6.3.14); K02111
F-type H+-transporting ATPase subunit alpha [EC:3.6.3.14]
Length=513
Score = 37.0 bits (84), Expect = 0.017, Method: Compositional matrix adjust.
Identities = 16/54 (29%), Positives = 33/54 (61%), Gaps = 1/54 (1%)
Query 72 ISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLLGLGNVRAGELVEL 125
++ +E++++++QRI+ + E G + SV DG+ R+ GL + GE++ L
Sbjct 3 LNSTEISELIKQRIAQFNVVSEAHNE-GTIVSVSDGVIRIHGLADCMQGEMISL 55
> mmu:104831 Ptpn23, AI462446, PTP-TD14; protein tyrosine phosphatase,
non-receptor type 23 (EC:3.1.3.48); K01104 protein-tyrosine
phosphatase [EC:3.1.3.48]
Length=1692
Score = 32.0 bits (71), Expect = 0.46, Method: Compositional matrix adjust.
Identities = 17/66 (25%), Positives = 32/66 (48%), Gaps = 3/66 (4%)
Query 53 QQQQRRLLHTSDLRAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDGIARLL 112
+QQ R L+ D+ A+ T SEM K+ E+++ + + + + + D + R L
Sbjct 576 EQQLRELIQKDDITASLVTTDHSEMKKLFEEQLKKYDQLKVY---LEQNLAAQDNVLRAL 632
Query 113 GLGNVR 118
NV+
Sbjct 633 TEANVQ 638
> hsa:25930 PTPN23, DKFZp564F0923, HD-PTP, HDPTP, KIAA1471, PTP-TD14;
protein tyrosine phosphatase, non-receptor type 23 (EC:3.1.3.48);
K01104 protein-tyrosine phosphatase [EC:3.1.3.48]
Length=1636
Score = 31.6 bits (70), Expect = 0.70, Method: Compositional matrix adjust.
Identities = 12/36 (33%), Positives = 21/36 (58%), Gaps = 0/36 (0%)
Query 53 QQQQRRLLHTSDLRAAATTISPSEMAKILEQRISGW 88
+QQ R L+ D+ A+ T SEM K+ E+++ +
Sbjct 574 EQQLRELIQKDDITASLVTTDHSEMKKLFEEQLKKY 609
> hsa:152992 METTL19, C4orf23, FLJ12891, FLJ35725; methyltransferase
like 19
Length=757
Score = 30.8 bits (68), Expect = 1.1, Method: Compositional matrix adjust.
Identities = 19/43 (44%), Positives = 23/43 (53%), Gaps = 2/43 (4%)
Query 18 LIRPLSSAAAACAARLSARLPAA--AAAAAGPAAAAGQQQQRR 58
L RP + C ARL AR AA A A GP +AG +Q+ R
Sbjct 28 LERPQVANKRLCGARLEARWSAALPCAEARGPGTSAGSEQKER 70
> cel:C50D2.7 hypothetical protein; K08074 ADP-dependent glucokinase
[EC:2.7.1.147]
Length=502
Score = 30.4 bits (67), Expect = 1.6, Method: Composition-based stats.
Identities = 22/73 (30%), Positives = 30/73 (41%), Gaps = 8/73 (10%)
Query 26 AAACAARLSARLPAAAAAAAGPAAAAGQQQQRRLLHTSDLRAAATTISPSEMAKILEQR- 84
AA A R++A P+ GP Q LLH S R +T I E+ ILE +
Sbjct 152 AALMADRIAANFPSTEVYLVGPIGPRSQA----LLHPSVKRTNSTRILKDELHVILEYKQ 207
Query 85 ---ISGWTTQQST 94
+ W S+
Sbjct 208 GEILGDWVAPSSS 220
> cel:Y11D7A.14 hypothetical protein
Length=1464
Score = 30.0 bits (66), Expect = 1.7, Method: Composition-based stats.
Identities = 20/65 (30%), Positives = 32/65 (49%), Gaps = 10/65 (15%)
Query 68 AATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGDG-----IARLLGLGNVRAGEL 122
A T S S MAKIL +R+ GW ++ FSV D ++R + + ++ E+
Sbjct 401 AKTLSSASAMAKILYERLFGWIVKRCNDA-----FSVDDTESTCRLSRFIAVLDIAGFEI 455
Query 123 VELQS 127
+E S
Sbjct 456 IEKNS 460
> xla:447469 usp38, MGC81730; ubiquitin specific peptidase 38
(EC:3.1.2.15); K11854 ubiquitin carboxyl-terminal hydrolase
35/38 [EC:3.1.2.15]
Length=1037
Score = 28.1 bits (61), Expect = 7.7, Method: Composition-based stats.
Identities = 19/58 (32%), Positives = 26/58 (44%), Gaps = 1/58 (1%)
Query 60 LHTSDLRAAATTISPSEMAKILEQRISGWTTQQSTAGEVGRVFSVGD-GIARLLGLGN 116
L+ L A + PSE L S WT+Q ST + R+ + G L+ LGN
Sbjct 394 LYEPILEALKDLLKPSEEKMKLTLNQSAWTSQSSTLASLPRLSGKSETGKTGLINLGN 451
> tgo:TGME49_052060 hypothetical protein
Length=438
Score = 27.7 bits (60), Expect = 9.0, Method: Compositional matrix adjust.
Identities = 26/80 (32%), Positives = 42/80 (52%), Gaps = 3/80 (3%)
Query 38 PAAAAAAAGPAAAAGQQQQRRLLHTSDLRAAATTISPSEMAKILEQRISGWTTQ-QSTAG 96
PA + ++AG AA Q ++RRLL +AAA ++P +M +ISG +TQ S
Sbjct 267 PAGSGSSAG-VPAAPQPKKRRLLDFVSAQAAAHQLAP-QMHTPPPGQISGTSTQIPSEHL 324
Query 97 EVGRVFSVGDGIARLLGLGN 116
+G + + G + L G+
Sbjct 325 PLGPLEASGPSLTSFLSTGH 344
Lambda K H
0.318 0.128 0.353
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 2099897216
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
Posted date: Sep 17, 2011 2:57 PM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40