bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
164,496 sequences; 82,071,388 total letters
Query= Emax_1973_orf2
Length=176
Score E
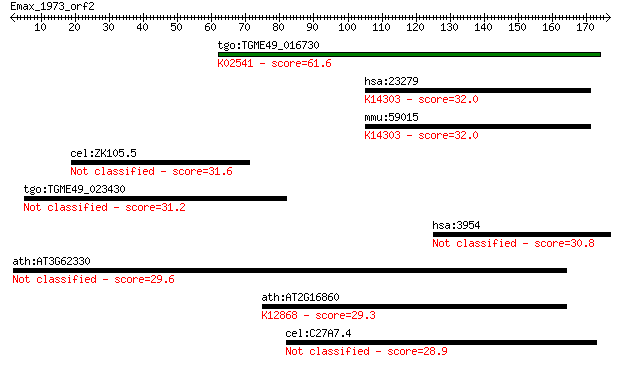
Sequences producing significant alignments: (Bits) Value
tgo:TGME49_016730 DNA replication licensing factor, putative ;... 61.6 1e-09
hsa:23279 NUP160, DKFZp686M14102, MGC150678, MGC150679; nucleo... 32.0 0.99
mmu:59015 Nup160, 160kDa, 2810011M03Rik, AA414952, AU020188, G... 32.0 1.1
cel:ZK105.5 hypothetical protein 31.6 1.6
tgo:TGME49_023430 hypothetical protein 31.2 2.1
hsa:3954 LETM1; leucine zipper-EF-hand containing transmembran... 30.8 2.7
ath:AT3G62330 zinc knuckle (CCHC-type) family protein 29.6 5.2
ath:AT2G16860 GCIP-interacting family protein; K12868 pre-mRNA... 29.3 6.7
cel:C27A7.4 che-11; abnormal CHEmotaxis family member (che-11) 28.9 8.2
> tgo:TGME49_016730 DNA replication licensing factor, putative
; K02541 minichromosome maintenance protein 3
Length=963
Score = 61.6 bits (148), Expect = 1e-09, Method: Compositional matrix adjust.
Identities = 45/144 (31%), Positives = 69/144 (47%), Gaps = 33/144 (22%)
Query 62 MASYIDDEID----GSLGGGQTEGN-------------MMGATSIDMTQAAPRRAAAAAA 104
MASY+ D+ D G+ G TEG MMG T ++ + RR A
Sbjct 1 MASYVTDDFDRPEPGAPGADTTEGGEEDMTMTMSGHPGMMGET-LEFQTNSMRRGGATNK 59
Query 105 ATGTATE---------------AEIDWKGEALMEQQMGHESNREKTATVRALTEAFQQTL 149
G T A ++++ E +++Q+G ESN E+ ++ L+ AF++ L
Sbjct 60 QGGLRTRVISTMSEGGLESAEGALVNYEEEVKLQRQLGEESNIERGRRMKDLSNAFKRDL 119
Query 150 AAIPGLSQQLDLLQQNAAAICLGM 173
++P + QQLDLLQ AAAI G+
Sbjct 120 ESVPAVRQQLDLLQNRAAAIVSGL 143
> hsa:23279 NUP160, DKFZp686M14102, MGC150678, MGC150679; nucleoporin
160kDa; K14303 nuclear pore complex protein Nup160
Length=1436
Score = 32.0 bits (71), Expect = 0.99, Method: Composition-based stats.
Identities = 23/73 (31%), Positives = 32/73 (43%), Gaps = 20/73 (27%)
Query 105 ATGTATEAEIDWKGEALMEQ-------QMGHESNREKTATVRALTEAFQQTLAAIPGLSQ 157
AT TEA DWK +A + +GH S +A++ L IP S+
Sbjct 983 ATSAITEAGDDWKSQATLRTCIFKHHLDLGHNS------------QAYE-ALTQIPDSSR 1029
Query 158 QLDLLQQNAAAIC 170
QLD L+Q +C
Sbjct 1030 QLDCLRQLVVVLC 1042
> mmu:59015 Nup160, 160kDa, 2810011M03Rik, AA414952, AU020188,
Gtl-13, Gtl1-13, Gtl13, KIAA0197, mKIAA0197; nucleoporin 160;
K14303 nuclear pore complex protein Nup160
Length=1402
Score = 32.0 bits (71), Expect = 1.1, Method: Composition-based stats.
Identities = 23/73 (31%), Positives = 32/73 (43%), Gaps = 20/73 (27%)
Query 105 ATGTATEAEIDWKGEALMEQ-------QMGHESNREKTATVRALTEAFQQTLAAIPGLSQ 157
AT TEA DWK +A + +GH S +A++ L IP S+
Sbjct 949 ATSAITEAGDDWKSQATLRTCIFKHHLDLGHNS------------QAYE-ALTQIPDSSR 995
Query 158 QLDLLQQNAAAIC 170
QLD L+Q +C
Sbjct 996 QLDCLRQLVVVLC 1008
> cel:ZK105.5 hypothetical protein
Length=569
Score = 31.6 bits (70), Expect = 1.6, Method: Composition-based stats.
Identities = 17/52 (32%), Positives = 26/52 (50%), Gaps = 3/52 (5%)
Query 19 TPAGSASALDPYCIFRNSTSCCSDGITDHIRNRSRSSSWRGKKMASYIDDEI 70
TP S DPY F + S IT+++ N+ + W KKM+ Y +E+
Sbjct 163 TPTSETSCKDPYYAFESCRCRQSYDITENLVNKYK---WADKKMSIYKQEEV 211
> tgo:TGME49_023430 hypothetical protein
Length=716
Score = 31.2 bits (69), Expect = 2.1, Method: Composition-based stats.
Identities = 25/84 (29%), Positives = 37/84 (44%), Gaps = 7/84 (8%)
Query 5 QRQGQQQLQHQPTETPAGSASALDPY---CIFRNSTSCCSDGIT--DHIRNRSRSSSWRG 59
Q++ Q H P + + L + C C+DG D +N + S+SWRG
Sbjct 143 QQRNQAFFVHGPDDGAHANTIVLGVWAAQCKNVEDVCLCTDGNCGFDRTKNANCSASWRG 202
Query 60 KKMASYI--DDEIDGSLGGGQTEG 81
K A+Y DD + G G G + G
Sbjct 203 KPGATYCQSDDWLGGKDGTGWSCG 226
> hsa:3954 LETM1; leucine zipper-EF-hand containing transmembrane
protein 1
Length=739
Score = 30.8 bits (68), Expect = 2.7, Method: Composition-based stats.
Identities = 19/53 (35%), Positives = 27/53 (50%), Gaps = 1/53 (1%)
Query 125 QMGHESNREKTATVRALTEAFQQTLAAIPGLSQQLDLLQQNAA-AICLGMPAG 176
+ G E E++ + LT+ QQ + I GL QL++ QQ A GMP G
Sbjct 592 KTGEEKYVEESKASKRLTKRVQQMIGQIDGLISQLEMDQQAGKLAPANGMPTG 644
> ath:AT3G62330 zinc knuckle (CCHC-type) family protein
Length=479
Score = 29.6 bits (65), Expect = 5.2, Method: Compositional matrix adjust.
Identities = 40/168 (23%), Positives = 68/168 (40%), Gaps = 20/168 (11%)
Query 2 VDHQRQGQQQLQHQPTETPAGSASALDPYCIFRNSTSCCSDGITDHIRNRSRSSSWRGKK 61
VD++ + + +++ P GS A ST S G R R S W K
Sbjct 237 VDNRFREETRVRENQRNVPRGSPQAYGSDRARSRSTHSKSPG-------RPRYSGW--DK 287
Query 62 MASYIDDEIDGSLGGGQTEGNMMGATSIDMTQAAPRRAAAAAAATGTATEAEIDWKGEAL 121
E+ G + M G++ I ++ R T E E+++ +AL
Sbjct 288 PYDRQKPEVSGYRSERWDQERMGGSSDIQVSHQFER-----PPFPQTLEELELEYTRDAL 342
Query 122 -MEQQMGHESNREKTA---TVRALTEAFQQTLAAIPGLS--QQLDLLQ 163
+E++ E + E T+R L E++ + LA + G++ Q D LQ
Sbjct 343 ELEKKRDKEEDEENNKHRETIRELRESYMKKLAGLRGMNAKQWDDFLQ 390
> ath:AT2G16860 GCIP-interacting family protein; K12868 pre-mRNA-splicing
factor SYF2
Length=298
Score = 29.3 bits (64), Expect = 6.7, Method: Compositional matrix adjust.
Identities = 21/89 (23%), Positives = 39/89 (43%), Gaps = 0/89 (0%)
Query 75 GGGQTEGNMMGATSIDMTQAAPRRAAAAAAATGTATEAEIDWKGEALMEQQMGHESNREK 134
G + G ++ A +DMTQA AA + E E G + Q+ + + +++
Sbjct 132 GRKKKIGKLLDANGLDMTQAYMLDTQEAAESKYKKWEKEPTPAGWDVFNQKTLYNAYKKR 191
Query 135 TATVRALTEAFQQTLAAIPGLSQQLDLLQ 163
T ++ E + + AA P ++ LQ
Sbjct 192 TKNIQVDLEEYNRMRAADPEFYREASSLQ 220
> cel:C27A7.4 che-11; abnormal CHEmotaxis family member (che-11)
Length=1437
Score = 28.9 bits (63), Expect = 8.2, Method: Composition-based stats.
Identities = 26/91 (28%), Positives = 43/91 (47%), Gaps = 7/91 (7%)
Query 82 NMMGATSIDMTQAAPRRAAAAAAATGTATEAEIDWKGEALMEQQMGHESNREKTATVRAL 141
N+ +T +D +Q + R + + TE ++ GE + +M R K A ++
Sbjct 349 NLSPSTHVD-SQVSLIRWSPILSTAALITEEDLVLIGENSLTVKM-----RGKMAAIQTS 402
Query 142 TEAFQQTLAAIPGLSQQLDLLQQNAAAICLG 172
+ +F L A G+SQ L L +A ICLG
Sbjct 403 SNSFT-LLHATSGVSQDLKLSIPSAKGICLG 432
Lambda K H
0.312 0.124 0.357
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 4600750868
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
Posted date: Sep 17, 2011 11:19 AM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40