bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
164,496 sequences; 82,071,388 total letters
Query= Emax_1655_orf1
Length=124
Score E
Sequences producing significant alignments: (Bits) Value
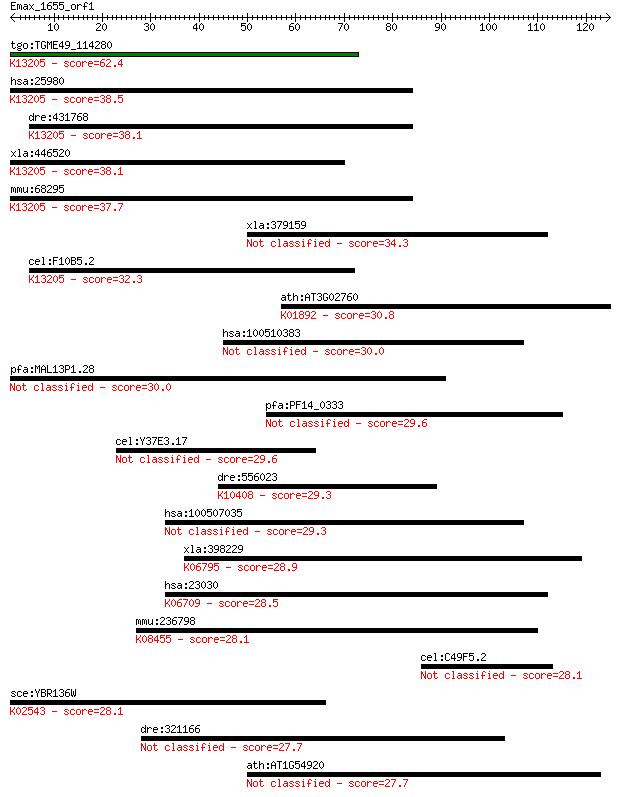
tgo:TGME49_114280 hypothetical protein ; K13205 A1 cistron-spl... 62.4 3e-10
hsa:25980 C20orf4, DKFZp564N1363, bA234K24.2; chromosome 20 op... 38.5 0.005
dre:431768 im:7153138; zgc:92018; K13205 A1 cistron-splicing f... 38.1 0.006
xla:446520 c20orf4, MGC80226; chromosome 20 open reading frame... 38.1 0.007
mmu:68295 0610011L14Rik; RIKEN cDNA 0610011L14 gene; K13205 A1... 37.7 0.009
xla:379159 MGC130823; hypothetical protein MGC52890 34.3 0.10
cel:F10B5.2 hypothetical protein; K13205 A1 cistron-splicing f... 32.3 0.39
ath:AT3G02760 ATP binding / aminoacyl-tRNA ligase/ histidine-t... 30.8 1.2
hsa:100510383 hypothetical protein LOC100510383 30.0 1.7
pfa:MAL13P1.28 conserved Plasmodium protein, unknown function 30.0 2.1
pfa:PF14_0333 conserved Plasmodium protein, unknown function 29.6 2.5
cel:Y37E3.17 hypothetical protein 29.6 2.5
dre:556023 dynein, axonemal, heavy chain 2-like; K10408 dynein... 29.3 3.3
hsa:100507035 Golgin subfamily A member 8-like protein 1-like 29.3 3.6
xla:398229 bcan, Xbcan; brevican; K06795 brevican 28.9 4.7
hsa:23030 KDM4B, FLJ44906, JMJD2B, KIAA0876; lysine (K)-specif... 28.5 5.2
mmu:236798 Gpr112, Gm367, PGR17; G protein-coupled receptor 11... 28.1 7.2
cel:C49F5.2 set-6; SET (trithorax/polycomb) domain containing ... 28.1 7.3
sce:YBR136W MEC1, ESR1, SAD3; Genome integrity checkpoint prot... 28.1 7.7
dre:321166 apobl, cb235, hm:zeh1207, sb:cb235, si:ch211-219i10... 27.7 8.6
ath:AT1G54920 hypothetical protein 27.7 9.1
> tgo:TGME49_114280 hypothetical protein ; K13205 A1 cistron-splicing
factor AAR2
Length=653
Score = 62.4 bits (150), Expect = 3e-10, Method: Composition-based stats.
Identities = 28/72 (38%), Positives = 42/72 (58%), Gaps = 0/72 (0%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
L H F W++L+ LL + RA+ LP +Y FL + QL Q PDD++ L + SF
Sbjct 475 LGHRFDGFEQWKQLLLLLCHCERAIRFLPEMYVSFLRLLHCQLEQCPDDLLSDSLLQKSF 534
Query 61 LSSSCASLLENC 72
L SS ++L++ C
Sbjct 535 LFSSASALVDTC 546
> hsa:25980 C20orf4, DKFZp564N1363, bA234K24.2; chromosome 20
open reading frame 4; K13205 A1 cistron-splicing factor AAR2
Length=384
Score = 38.5 bits (88), Expect = 0.005, Method: Composition-based stats.
Identities = 24/85 (28%), Positives = 41/85 (48%), Gaps = 2/85 (2%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
L + + + HW+ L+ LL + A+ LY + Y QL + P D + + + +F
Sbjct 264 LGNVYEAFEHWKRLLNLLCRSEAAMMKHHTLYINLISILYHQLGEIPADFFVDIVSQDNF 323
Query 61 LSSSCASLLEN-CSY-IDRQLAKKA 83
L+S+ + CS +D L KKA
Sbjct 324 LTSTLQVFFSSACSIAVDATLRKKA 348
> dre:431768 im:7153138; zgc:92018; K13205 A1 cistron-splicing
factor AAR2
Length=384
Score = 38.1 bits (87), Expect = 0.006, Method: Composition-based stats.
Identities = 22/81 (27%), Positives = 38/81 (46%), Gaps = 2/81 (2%)
Query 5 FPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSFLSSS 64
+ + HW+ L+ LL + A+ LY + Y QL + P D L + + +FL+S+
Sbjct 268 YEAFEHWKSLLALLCRSEEAMKDRKELYLGLIAVLYHQLGEIPPDFFLDIVSQSNFLTST 327
Query 65 CASLLENCSY--IDRQLAKKA 83
+ S +D L K+A
Sbjct 328 LQDFFQFASSPGVDSTLRKRA 348
> xla:446520 c20orf4, MGC80226; chromosome 20 open reading frame
4; K13205 A1 cistron-splicing factor AAR2
Length=388
Score = 38.1 bits (87), Expect = 0.007, Method: Composition-based stats.
Identities = 20/69 (28%), Positives = 33/69 (47%), Gaps = 0/69 (0%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
L + + + W+ L+ LL A P LY K + Y QL+Q P D + + + +F
Sbjct 258 LGNVYEAFEQWKSLLNLLCRAETFSLQHPELYIKVISVLYHQLAQVPTDFFVDIVSQNNF 317
Query 61 LSSSCASLL 69
L+S+ L
Sbjct 318 LTSTLQVLF 326
> mmu:68295 0610011L14Rik; RIKEN cDNA 0610011L14 gene; K13205
A1 cistron-splicing factor AAR2
Length=384
Score = 37.7 bits (86), Expect = 0.009, Method: Composition-based stats.
Identities = 23/85 (27%), Positives = 41/85 (48%), Gaps = 2/85 (2%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
L + + + HW+ L+ LL + A+ LY + Y QL + P D + + + +F
Sbjct 264 LGNVYEAFEHWKRLLNLLCRSESAMGKYHALYISLISILYHQLGEIPADFFVDIVSQDNF 323
Query 61 LSSSCASLLEN-CSY-IDRQLAKKA 83
L+S+ + CS ++ L KKA
Sbjct 324 LTSTLQVFFSSACSIAVEATLRKKA 348
> xla:379159 MGC130823; hypothetical protein MGC52890
Length=330
Score = 34.3 bits (77), Expect = 0.10, Method: Compositional matrix adjust.
Identities = 24/68 (35%), Positives = 34/68 (50%), Gaps = 6/68 (8%)
Query 50 IILGPLEEGS--FLSSSCASLLE----NCSYIDRQLAKKALIQQITNNIQEKKQKKIQNI 103
II G +E G+ +L+ CA+LL+ NC +D AL Q NN++ + I
Sbjct 92 IIHGFIERGTDKWLTHMCANLLQVEDVNCLCVDWAGGSYALFSQAANNVRVVGAEVAHFI 151
Query 104 HQLSNKKG 111
LSNK G
Sbjct 152 QLLSNKYG 159
> cel:F10B5.2 hypothetical protein; K13205 A1 cistron-splicing
factor AAR2
Length=357
Score = 32.3 bits (72), Expect = 0.39, Method: Compositional matrix adjust.
Identities = 15/67 (22%), Positives = 31/67 (46%), Gaps = 0/67 (0%)
Query 5 FPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSFLSSS 64
F W+ +I L+S ++ L+ F+ + QL + P D + + +FL+++
Sbjct 258 FEGFEQWKRIIHLMSCCPNSLGSEKELFMSFIRVLFFQLKECPTDFFVDIVSRDNFLTTT 317
Query 65 CASLLEN 71
+ L N
Sbjct 318 LSMLFAN 324
> ath:AT3G02760 ATP binding / aminoacyl-tRNA ligase/ histidine-tRNA
ligase/ nucleotide binding (EC:6.1.1.21); K01892 histidyl-tRNA
synthetase [EC:6.1.1.21]
Length=883
Score = 30.8 bits (68), Expect = 1.2, Method: Composition-based stats.
Identities = 22/72 (30%), Positives = 34/72 (47%), Gaps = 7/72 (9%)
Query 57 EGSFLSSSCASLLENCSYIDRQLAKKALIQQITNNIQ----EKKQKKIQNIHQLSNKKGK 112
E L SCASL+ CS ID K + QI +++ E + I + L + G
Sbjct 120 ENIVLEKSCASLIGICSIIDH---KSTTLSQIVDSVAALSCEVTKADIASFSLLDSGDGN 176
Query 113 GNKNNIIIQEDM 124
G+K+ I + D+
Sbjct 177 GDKDVIGVAGDL 188
> hsa:100510383 hypothetical protein LOC100510383
Length=389
Score = 30.0 bits (66), Expect = 1.7, Method: Composition-based stats.
Identities = 22/63 (34%), Positives = 30/63 (47%), Gaps = 1/63 (1%)
Query 45 QSPDDIILGPLEEGSFLSSSCASLLENCSYIDRQLAKKAL-IQQITNNIQEKKQKKIQNI 103
QSP D G EG S++ L C L +++ I Q+ N I+ KQ+K Q
Sbjct 323 QSPGDSATGIHGEGPTSSATLKDLESPCQEPAVVLNPRSVKISQLKNTIKSLKQQKKQVE 382
Query 104 HQL 106
HQL
Sbjct 383 HQL 385
> pfa:MAL13P1.28 conserved Plasmodium protein, unknown function
Length=609
Score = 30.0 bits (66), Expect = 2.1, Method: Composition-based stats.
Identities = 21/90 (23%), Positives = 41/90 (45%), Gaps = 2/90 (2%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
L +++ S + WR+L +L++N+ V + + L QLS +D + SF
Sbjct 490 LGYNYQSFIQWRKLFELMTNSSYLVTNSTNFFEEVLKAVCIQLSYLSEDFFDN--SDNSF 547
Query 61 LSSSCASLLENCSYIDRQLAKKALIQQITN 90
+ ++ E + ID ++ K + I N
Sbjct 548 ILFGIYNIYEIINNIDEKIRPKNITTLIDN 577
> pfa:PF14_0333 conserved Plasmodium protein, unknown function
Length=359
Score = 29.6 bits (65), Expect = 2.5, Method: Compositional matrix adjust.
Identities = 20/61 (32%), Positives = 32/61 (52%), Gaps = 7/61 (11%)
Query 54 PLEEGSFLSSSCASLLENCSYIDRQLAKKALIQQITNNIQEKKQKKIQNIHQLSNKKGKG 113
P ++GSFL +EN I KK +IQ++ N + + Q ++ N+ + NKKG
Sbjct 271 PQKDGSFL-------IENVDPIKYNNKKKLIIQELLNEHENRFQSQVSNLKKKINKKGYT 323
Query 114 N 114
N
Sbjct 324 N 324
> cel:Y37E3.17 hypothetical protein
Length=837
Score = 29.6 bits (65), Expect = 2.5, Method: Composition-based stats.
Identities = 15/42 (35%), Positives = 25/42 (59%), Gaps = 1/42 (2%)
Query 23 RAVYLLPGLYGKFLDTFYAQLSQSPDDI-ILGPLEEGSFLSS 63
RA L+PGL G +D A S +PD ++GP ++ ++S+
Sbjct 339 RACDLIPGLQGSKVDARAAVFSMTPDGYPLVGPYDKNYWMST 380
> dre:556023 dynein, axonemal, heavy chain 2-like; K10408 dynein
heavy chain, axonemal
Length=4424
Score = 29.3 bits (64), Expect = 3.3, Method: Composition-based stats.
Identities = 23/49 (46%), Positives = 26/49 (53%), Gaps = 8/49 (16%)
Query 44 SQSPDDI--ILGPLEEGSFLSSSCASLLENCSYIDRQLAKKAL--IQQI 88
+QS DI LGPLEE SF S CA L I QL K + IQ+I
Sbjct 259 AQSVSDIGDTLGPLEEISFWKSRCADL----DSISTQLQKPGVRHIQEI 303
> hsa:100507035 Golgin subfamily A member 8-like protein 1-like
Length=123
Score = 29.3 bits (64), Expect = 3.6, Method: Compositional matrix adjust.
Identities = 24/75 (32%), Positives = 34/75 (45%), Gaps = 1/75 (1%)
Query 33 GKFLDTFYAQLSQSPDDIILGPLEEGSFLSSSCASLLENCSYIDRQLAKKAL-IQQITNN 91
G +T + QSP D G EG S++ L C L +++ I Q+ N
Sbjct 45 GSIPETATSGGCQSPGDSATGIHGEGPTSSATLKDLESPCQEPAVVLNPRSVKISQLKNT 104
Query 92 IQEKKQKKIQNIHQL 106
I+ KQ+K Q HQL
Sbjct 105 IKSLKQQKKQVEHQL 119
> xla:398229 bcan, Xbcan; brevican; K06795 brevican
Length=1152
Score = 28.9 bits (63), Expect = 4.7, Method: Composition-based stats.
Identities = 18/82 (21%), Positives = 36/82 (43%), Gaps = 10/82 (12%)
Query 37 DTFYAQLSQSPDDIILGPLEEGSFLSSSCASLLENCSYIDRQLAKKALIQQITNNIQEKK 96
D FY ++ PD+ F S +L + + + + Q+ NN EK+
Sbjct 535 DYFYDNITNLPDE----------FGSEESETLNQTFNTTTQSPQRSLETDQLMNNHGEKE 584
Query 97 QKKIQNIHQLSNKKGKGNKNNI 118
+++ NI +NK+ + + N +
Sbjct 585 NEEVYNISVTTNKQSEASSNPV 606
> hsa:23030 KDM4B, FLJ44906, JMJD2B, KIAA0876; lysine (K)-specific
demethylase 4B; K06709 jumonji domain-containing protein
2 [EC:1.14.11.-]
Length=1096
Score = 28.5 bits (62), Expect = 5.2, Method: Composition-based stats.
Identities = 22/79 (27%), Positives = 34/79 (43%), Gaps = 12/79 (15%)
Query 33 GKFLDTFYAQLSQSPDDIILGPLEEGSFLSSSCASLLENCSYIDRQLAKKALIQQITNNI 92
G + D Y + S D + LGP EG L + D L K I +T++I
Sbjct 955 GSYSDNLYPESITSRDCVQLGPPSEGE---------LVELRWTDGNLYKAKFISSVTSHI 1005
Query 93 QEKKQKKIQNIHQLSNKKG 111
Q + ++ QL+ K+G
Sbjct 1006 Y---QVEFEDGSQLTVKRG 1021
> mmu:236798 Gpr112, Gm367, PGR17; G protein-coupled receptor
112; K08455 G protein-coupled receptor 112
Length=3056
Score = 28.1 bits (61), Expect = 7.2, Method: Composition-based stats.
Identities = 30/95 (31%), Positives = 44/95 (46%), Gaps = 15/95 (15%)
Query 27 LLPGLYGKFLDTFYAQLS-QSPDDI---ILG------PL--EEGSFLSSSCASLLENCSY 74
LL GL K +D +S ++ DD+ IL PL EE + S A + NC
Sbjct 2381 LLQGLPDKIVDLANITISDENADDVAEHILNLVNESPPLDEEETKIIVSKVADI-SNCDE 2439
Query 75 IDRQLAKKALIQQITNNIQEKKQKKIQNIHQLSNK 109
I L + +I QI N + EK+ N+ +SN+
Sbjct 2440 ISINLTQ--IILQILNTVTEKQSDSASNLPPVSNE 2472
> cel:C49F5.2 set-6; SET (trithorax/polycomb) domain containing
family member (set-6)
Length=708
Score = 28.1 bits (61), Expect = 7.3, Method: Composition-based stats.
Identities = 9/27 (33%), Positives = 21/27 (77%), Gaps = 0/27 (0%)
Query 86 QQITNNIQEKKQKKIQNIHQLSNKKGK 112
+ I+NN+ EK+++ +++ H+L +K+ K
Sbjct 89 RAISNNLTEKRRRSVESFHRLHSKERK 115
> sce:YBR136W MEC1, ESR1, SAD3; Genome integrity checkpoint protein
and PI kinase superfamily member; signal transducer required
for cell cycle arrest and transcriptional responses prompted
by damaged or unreplicated DNA; monitors and participates
in meiotic recombination (EC:2.7.11.1); K02543 cell cycle
checkpoint protein MEC1
Length=2368
Score = 28.1 bits (61), Expect = 7.7, Method: Compositional matrix adjust.
Identities = 20/65 (30%), Positives = 31/65 (47%), Gaps = 5/65 (7%)
Query 1 LAHHFPSLLHWRELIQLLSNAGRAVYLLPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSF 60
LA +PS + W + SN+ + V L GK + Y Q SQ+P D++ L+
Sbjct 1926 LAVEYPSHILWYITALVNSNSSKRV-----LRGKHILEKYRQHSQNPHDLVSSALDLTKA 1980
Query 61 LSSSC 65
L+ C
Sbjct 1981 LTRVC 1985
> dre:321166 apobl, cb235, hm:zeh1207, sb:cb235, si:ch211-219i10.1,
wu:fb39c11, wu:fc51e12, zeh1207; apolipoprotein B, like
Length=3730
Score = 27.7 bits (60), Expect = 8.6, Method: Composition-based stats.
Identities = 22/76 (28%), Positives = 39/76 (51%), Gaps = 8/76 (10%)
Query 28 LPGLYGKFLDTFYAQLSQSPDDIILGPLEEGSFLSSSCASLLENCSYIDRQLAKKALIQQ 87
L G+ G F DT ++ + D + G + S +L N I Q A ++++++
Sbjct 662 LIGIDGFFRDTMQKTINYAADKVPRG----NDIMQSMFPTLWNN---IKMQKAPQSIVKE 714
Query 88 ITNNIQEKKQK-KIQN 102
ITNN+ + QK K+Q+
Sbjct 715 ITNNVNKLIQKLKVQD 730
> ath:AT1G54920 hypothetical protein
Length=766
Score = 27.7 bits (60), Expect = 9.1, Method: Composition-based stats.
Identities = 24/80 (30%), Positives = 40/80 (50%), Gaps = 15/80 (18%)
Query 50 IILGPLEEGSF-------LSSSCASLLENCSYIDRQLAKKALIQQITNNIQEKKQKKIQN 102
I LG LE+ S +SS+CA L + + I +Q+ I N++QE+ K Q
Sbjct 693 INLGSLEQQSKYASTWFEISSTCAQELRHAASIWKQV--------IKNDVQEEILSKPQE 744
Query 103 IHQLSNKKGKGNKNNIIIQE 122
+ LS + + +KN+ I +
Sbjct 745 LCSLSGRNLQSSKNSESINQ 764
Lambda K H
0.318 0.135 0.386
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 2069971060
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
Posted date: Sep 17, 2011 11:19 AM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40