bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.30+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF
excluding environmental samples from WGS projects
49,011,213 sequences; 17,563,301,199 total letters
Query= Contig-1_CDS_annotation_glimmer3.pl_2_5
Length=133
Score E
Sequences producing significant alignments: (Bits) Value
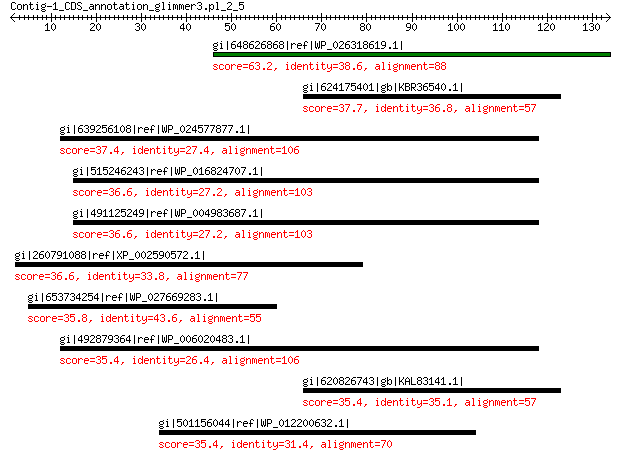
gi|648626868|ref|WP_026318619.1| hypothetical protein 63.2 3e-10
gi|624175401|gb|KBR36540.1| hypothetical protein X418_00653 37.7 1.1
gi|639256108|ref|WP_024577877.1| MULTISPECIES: TetR family trans... 37.4 1.7
gi|515246243|ref|WP_016824707.1| peptidase 36.6 2.8
gi|491125249|ref|WP_004983687.1| peptidase 36.6 2.8
gi|260791088|ref|XP_002590572.1| hypothetical protein BRAFLDRAFT... 36.6 3.2
gi|653734254|ref|WP_027669283.1| hypothetical protein 35.8 5.9
gi|492879364|ref|WP_006020483.1| MULTISPECIES: hypothetical protein 35.4 6.6
gi|620826743|gb|KAL83141.1| hypothetical protein AJ09_00840 35.4 7.1
gi|501156044|ref|WP_012200632.1| DNA polymerase III subunit alpha 35.4 7.6
>gi|648626868|ref|WP_026318619.1| hypothetical protein [Alistipes onderdonkii]
Length=101
Score = 63.2 bits (152), Expect = 3e-10, Method: Compositional matrix adjust.
Identities = 34/88 (39%), Positives = 48/88 (55%), Gaps = 7/88 (8%)
Query 46 FIVGARSMNEILEEYYTFGFLSCDTQAVRGDSAYDEIQPSGKDASFLSTDPSSDFSLDKF 105
I G +S+ + L+EY+ G CD YDE S DPS+DFSLDKF
Sbjct 17 LIHGVKSLRQGLQEYWETGVYPCDPVQRNSGEYYDE-------DSTTQVDPSTDFSLDKF 69
Query 106 ERIERIAECVGETSAERHKEELGKQNDK 133
ER+E+I E V E ++H+++LG D+
Sbjct 70 ERLEKIVEVVSERERKKHEDDLGVTPDE 97
>gi|624175401|gb|KBR36540.1| hypothetical protein X418_00653 [Mycobacterium tuberculosis XTB13-212]
Length=373
Score = 37.7 bits (86), Expect = 1.1, Method: Compositional matrix adjust.
Identities = 21/57 (37%), Positives = 32/57 (56%), Gaps = 2/57 (4%)
Query 66 LSCDTQAVRGDSAYDEIQPSGKDASFLSTDPSSDFSLDKFERIERIAECVGETSAER 122
L D ++RG A E P+ +D +F++ DP+S F+LD+ E + E V SA R
Sbjct 32 LGADAASIRG--AVSEQSPNYRDGAFVNLDPASMFTLDREELRLIVWELVARHSASR 86
>gi|639256108|ref|WP_024577877.1| MULTISPECIES: TetR family transcriptional regulator [Afipia]
Length=247
Score = 37.4 bits (85), Expect = 1.7, Method: Compositional matrix adjust.
Identities = 29/113 (26%), Positives = 51/113 (45%), Gaps = 15/113 (13%)
Query 12 GCLYSNLSQRVGIRSCADQHASYHVRNCVSQPDEFIVGARSMNEILEEYYTFGFLSCDTQ 71
G L+ +V + C +H + CV++ DE + GA + +E+LE GFL+ ++
Sbjct 57 GVLHYYFKDKVDLMMCCVRH---YKAVCVTRYDEVVAGAATYDELLE-----GFLAALSE 108
Query 72 AVRGDSAYDEIQPSGKDASFLSTDPSSD-FSLDK------FERIERIAECVGE 117
VR D+ + + S + +D +DK + + R AE GE
Sbjct 109 TVRNDAQMHRLWYDLRSQSLFEEEFRADVLEIDKSLEAMIWRIMSRFAELTGE 161
>gi|515246243|ref|WP_016824707.1| peptidase [Streptomyces viridosporus]
Length=523
Score = 36.6 bits (83), Expect = 2.8, Method: Composition-based stats.
Identities = 28/106 (26%), Positives = 46/106 (43%), Gaps = 12/106 (11%)
Query 15 YSNLSQRVGIRSCADQHASYHVRNCVSQPDEFIVGARSMNEILEEYYTFGFLSCDTQAVR 74
YSNL +CAD Y + EF R+ +E+ EY +G + C AV
Sbjct 376 YSNLVPANVSINCADDKPRYRTEDVERSLPEF----RAASELFGEYLAWGLIGCTDWAVP 431
Query 75 GDSAYDEIQPSGKDASFL---STDPSSDFSLDKFERIERIAECVGE 117
G + + E+ G + + DP++ +E R+A+ +GE
Sbjct 432 GAADHPEVSAPGAAPILVIGNTGDPAT-----PYEGARRMAQALGE 472
>gi|491125249|ref|WP_004983687.1| peptidase [Streptomyces ghanaensis]
gi|291340295|gb|EFE67251.1| peptidase [Streptomyces ghanaensis ATCC 14672]
Length=523
Score = 36.6 bits (83), Expect = 2.8, Method: Composition-based stats.
Identities = 28/106 (26%), Positives = 46/106 (43%), Gaps = 12/106 (11%)
Query 15 YSNLSQRVGIRSCADQHASYHVRNCVSQPDEFIVGARSMNEILEEYYTFGFLSCDTQAVR 74
YSNL +CAD Y + EF R+ +E+ EY +G + C AV
Sbjct 376 YSNLVPANVSINCADDKPRYRTEDVERSLPEF----RAASELFGEYLAWGLIGCTDWAVP 431
Query 75 GDSAYDEIQPSGKDASFL---STDPSSDFSLDKFERIERIAECVGE 117
G + + E+ G + + DP++ +E R+A+ +GE
Sbjct 432 GAADHPEVSAPGAAPILVIGNTGDPAT-----PYEGARRMAQALGE 472
>gi|260791088|ref|XP_002590572.1| hypothetical protein BRAFLDRAFT_123628 [Branchiostoma floridae]
gi|229275767|gb|EEN46583.1| hypothetical protein BRAFLDRAFT_123628 [Branchiostoma floridae]
Length=1136
Score = 36.6 bits (83), Expect = 3.2, Method: Compositional matrix adjust.
Identities = 26/89 (29%), Positives = 42/89 (47%), Gaps = 12/89 (13%)
Query 2 KTKQDYNPH---VGCLYSNLSQRVGIRSC-ADQHASYHVRNCVSQPDEFIVGARSMN--- 54
K ++++ PH V C +L Q R+ A+Q + + VS P + + A+S
Sbjct 192 KLQEEFTPHSKLVACALLSLHQVYPRRNLPAEQWRQAQMLSLVSAPGQMLNPAQSETMPC 251
Query 55 -----EILEEYYTFGFLSCDTQAVRGDSA 78
E +E++ FGFL C Q + DSA
Sbjct 252 EYLSVETMEKWIIFGFLLCHGQLTQSDSA 280
>gi|653734254|ref|WP_027669283.1| hypothetical protein [Rhizobium leguminosarum]
Length=256
Score = 35.8 bits (81), Expect = 5.9, Method: Compositional matrix adjust.
Identities = 24/74 (32%), Positives = 35/74 (47%), Gaps = 20/74 (27%)
Query 5 QDYNPHVGCLYSNLSQRVGI-------------------RSCADQHASYHVRNCVSQPDE 45
+D NP VGCLYS L Q +GI R A+ A+Y +R C S D+
Sbjct 182 RDINP-VGCLYSVLLQSIGIEGPFGGVAATYRAGDVPGFRRNAEMAAAYAIRECRSVVDK 240
Query 46 FIVGARSMNEILEE 59
+ + + E+L+E
Sbjct 241 KVQLEKCLREVLKE 254
>gi|492879364|ref|WP_006020483.1| MULTISPECIES: hypothetical protein [Afipia]
gi|410892009|gb|EKS39805.1| hypothetical protein HMPREF9695_01766 [Afipia broomeae ATCC 49717]
Length=259
Score = 35.4 bits (80), Expect = 6.6, Method: Compositional matrix adjust.
Identities = 28/113 (25%), Positives = 51/113 (45%), Gaps = 15/113 (13%)
Query 12 GCLYSNLSQRVGIRSCADQHASYHVRNCVSQPDEFIVGARSMNEILEEYYTFGFLSCDTQ 71
G L+ +V + C +H + CV++ DE + GA + +E+LE GFL+ ++
Sbjct 57 GVLHYYFKDKVDLMMCCVRH---YKAVCVTRYDEVVAGAATYDELLE-----GFLAALSE 108
Query 72 AVRGDSAYDEIQPSGKDASFLSTDPSSD-FSLDK------FERIERIAECVGE 117
VR D+ + + S + +D +DK + + R +E GE
Sbjct 109 TVRNDAQMHRLWYDLRSQSLFEEEFRADVLEIDKSLENMIWRIMSRFSELTGE 161
>gi|620826743|gb|KAL83141.1| hypothetical protein AJ09_00840 [Mycobacterium tuberculosis MD17749]
Length=372
Score = 35.4 bits (80), Expect = 7.1, Method: Compositional matrix adjust.
Identities = 20/57 (35%), Positives = 31/57 (54%), Gaps = 3/57 (5%)
Query 66 LSCDTQAVRGDSAYDEIQPSGKDASFLSTDPSSDFSLDKFERIERIAECVGETSAER 122
L D ++R A E P+ +D +F++ DP+S F+LD+ E + E V SA R
Sbjct 32 LGADAASIR---AVSEQSPNYRDGAFVNVDPASMFTLDREELRLIVWELVARHSASR 85
>gi|501156044|ref|WP_012200632.1| DNA polymerase III subunit alpha [Lachnoclostridium phytofermentans]
gi|160880751|ref|YP_001559719.1| DNA polymerase III subunit alpha [Lachnoclostridium phytofermentans
ISDg]
gi|160429417|gb|ABX42980.1| DNA polymerase III, alpha subunit [Lachnoclostridium phytofermentans
ISDg]
Length=1527
Score = 35.4 bits (80), Expect = 7.6, Method: Compositional matrix adjust.
Identities = 22/70 (31%), Positives = 38/70 (54%), Gaps = 5/70 (7%)
Query 34 YHVRNCVSQPDEFIVGARSMNEILEEYYTFGFLSCDTQAVRGDSAYDEIQPSGKDASFLS 93
YH++ S DE ++G+R++ EIL E+ +F C+ + +A +I K A FL+
Sbjct 543 YHIQKLTSITDEMVIGSRTIEEILPEFLSF----CEGCYLVAHNASFDIGFITKKAEFLN 598
Query 94 TDPSSDFSLD 103
P + S+D
Sbjct 599 I-PLTVTSVD 607
Lambda K H a alpha
0.315 0.131 0.383 0.792 4.96
Gapped
Lambda K H a alpha sigma
0.267 0.0410 0.140 1.90 42.6 43.6
Effective search space used: 432180497808